News & Updates
Variational Monte Carlo
Variational Monte Carlo is a method to evaluate the integral $\frac{ \int |\Psi(R)|^2 O(R) dR}{\int |\Psi(R)|^2} dR. $
To accomplish this, we use markov chain Monte Carlo. This works in the following manner:
- Choose a new trial location for the particles
- Evaluate the ratio squared of the wavefunctions for the new and old trial locations
- If the ratio squared is greater then some random number, then keep the new trial location, otherwise keep the old trial location.
Trial Wave functions for a hydrogen molecule
We will be writing a simple variational monte carlo code to calculate the bond length of a Hydrogen molecule.
Note: You will need to be analyzing some data in this section. Make
sure you are comfortable with the python statistical package we've supplied.
For sake of simplicity, this lab will focus on the hydrogen molecule. Since it has only two electrons (one spin up and one spin down), we need not be concerned with computing determinants. We will be optimizing two very simple trial wave functions $\Psi_1$ and $\Psi_2$. Let our molecule be centered at the origin with protons at $\pm L/2 \hat{x}$.
The first trial function is a simple bond-centered Gaussian with width controlled by the parameter $\alpha$, $\Psi(r_1,r_2;\alpha)=\exp[-\alpha(|r_1|^2+|r_2|^2)]$ where $\alpha$ is to be optimized variationally.
We will use as our data structure for storing the coordinates of the ions (electrons) to be a 2d numpy array. The first coordinate will be the particles and the second coordinate the coordinates. Therefore, to access the second particle, call coords[1] and to access the y element, call coords[1,1] (remember that in python, all arrays start counting from 0)
Building the WaveFunction
Let's start by building a test for our wave function. We will use $\alpha=0.5$. What should wave function do? It takes the position of the ions, the position of the electrons and gives a number. Let us then define our test as
import numpy
def WaveFunction1_test1(wavefunction):
coords=numpy.array([[1.0,0.5,0.3],[-0.2,0.1,-0.1]])
ions=numpy.array([[-0.7,0.0,0.0],[0.7,0.0,0.0]])
if numpy.abs(wavefunction(coords,ions)-0.496585)<1e-5:
return True
else:
return False
if (WaveFunction1_test1(psi)):
print "Wavefunction Test passed"
else:
print "Wavefunction Test Failed"
Now, you should write your wavefunction
def psi(coords,ions):
# your code here
return valremembering to put it somewhere in the file above the test. Useful things to know include
- import numpy and import math should be at the top of your file
- numpy.dot calculates dot products and
- math.sqrt does square roots.
After you have written your code, run your test and verify your wavefunction is working! (If you don't get this value, stop here and fix the problem. Building on a broken base will make things much harder to debug in the future).
Building a Monte Carlo Code
The next step in producing our Monte Carlo code, is to decide how we are going to move the particles. One natural (albeit not the best) approach to this is to simply move the particles in a small box around their current position. Let's start with this as our basic approach.
We are now ready to put everything together in our very first Monte Carlo code! In our first attempt, we will compute just one thing: the acceptance ratio of our moves. Observe that our basic process will be:
def VMC(WF,ions,numSteps):
R=numpy.zeros((2,2),float)
movesAttempted=0.0
movesAccepted=0.0
for step in xrange(0,numSteps):
for ptcl in xrange(0,len(R)):
a=5
# make your move for particle "ptcl"
# decide if you accepted or rejected
# updated movesAttempted and movesAccepted
# here you will compute other things in the next steps
print "Acceptance ratio: ", movesAccepted/movesAttempted
return To check if you've correctly put together your VMC code, you should find that if you are moving the particle in a box of length 1.5 (in each direction) around its current position you will get an acceptance ratio of approximately 0.323. Useful things to know include the fact that
- numpy.random.rand() will return a random number uniformly between 0 and 1
- to square a number do a**2 (the ^ will not work)
- Remember that you accept a move with the ratio of the wavefunctions squared
- You can add two arrays of the same size by using the +
Congratulations! If you are getting this number you've successfully produced your first (admittidly a bit useless) QMC code!
Computing the density
Note: If you are running low on time, feel free to skip this section
Of course, we want our QMC code to compute something. In this section, we will learn how to compute the density in the bond direction. In the next section, we will work on computing the energy. The energy will lead us into a variety of interesting questions about opimization, measuring bond-lengths, etc.
The key to computing the density in the x direction is to build up histogram. This is a good time to learn something about doing object oriented programing in python. In object oriented programming, you define a class. A class is essentially a group of functions and data that are put together so that the functions can access the data without them being part of it's arguments. Let's define a
class Histogram:
def Init(self,startPoint, endPoint,numPoints):
#here we will write code to set up our class.
def Add(self,data):
#add some code here
def Plot(self):
# let's just plot the histogram
- put import pylab at the top of your code
- pylab.plot(x,y) will prepare a plot and pylab.show() will show it.
- numpy.zeros(20,float) will create an array of floats with 20 zeros.
- int(x) will return an integer from a float.
- Remember that if you want your class to store an array to put your histogram in you would do self.myData=numpy.zeros(numPoints,float)
myHist=Histogram()
myHist.Init(-10,10,100)
sigma=3.0
randNums=numpy.random.randn(100000)*sigma
for r in randNums:
myHist.Add(r)
myHist.Plot()
This should produce a gaussian curve with a standard deviation of 3.0. Check to make sure this is the case before going
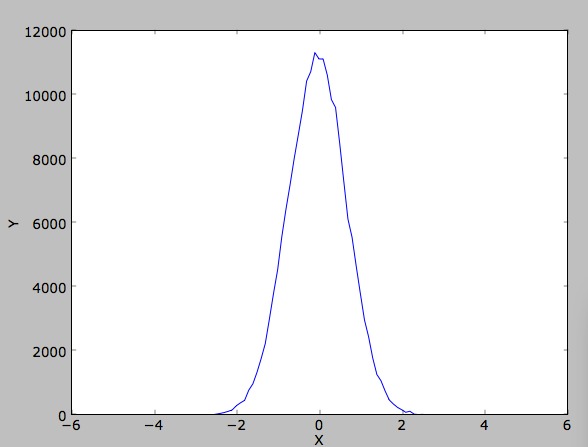
on! Now, use your histogram class to compute the density in the direction of the hydrogen molecules bond. You should
expect to see something like this (if you make enough VMC steps!):
Computing the local energy
def LaplacianPsiOverPsi(wavefunction,coords,ions):
#this should return the kinetic piece
# of the local energy
def LocalEnergy(wavefunction,coords,ions):
# this should return the local energy.
To verify that your code is correct, set your ions so that the bond length is 1.4 and set the coordinates via
R=numpy.zeros((2,3),float)
R[0]=[1.0,0.3,0.2]
R[1]=[2.0,-0.2,0.1]
Now add the calculation of the local energy to your VMC function. Have the VMC function return a list of energies. Once you have these energies, use the statitistical package to compute the energy and error as well as plotting the energies.
You should end up seeing a plot like:
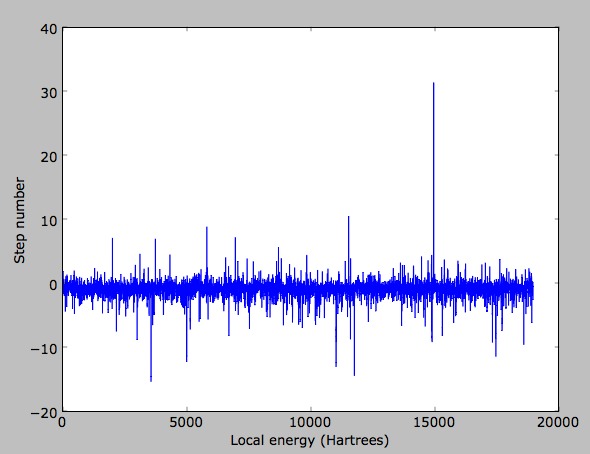
You should notice a couple things about this plot
- It is quite spiky.
- If you compute the statistics of your energy list, your answer is around -0.86.
- Your variance is large.
Classes all around
psi(coords,ions)
LaplacianPsiOverPsi(wavefunction,coords,ions)
LocalEnergy(wavefunction,coords,ions)
class WaveFunctionClass:
def psi(self,coords):
#stuff here
def LaplacianPsiOverPsi(self,coords):
#stuff here
def LocalEnergy(self , coords):
#stuff here
def SetIons(self,ions):
# stuff here
def SetAlpha(self,alpha):
#stuff here
Notice, that we don't have to pass around as many things (like alpha and the ions) and we still get away with not have ugly global variables. Of course, this means that you're going to have to play around with some of the function calls and such that you have in your code so far. Go ahead and make the changes in your code necessary to be using this class. (Looking through the section "Computing the density" is probably helpful now)
You should be able to test your modifications to your class with the following code (which should work if you have only the HistogramClass and the WaveFunctionClass in your file!):
import stats
wf=WaveFunctionClass()
wf.SetIons(numpy.array([[0.7,0.0,0.0],[-0.7,0.0,0.0]]))
wf.SetAlpha(0.5)
print "Psi should be 0.496585 when given coords as numpy.array([[1.0,0.5,0.3],[-0.2,0.1\
,-0.1]) and is ",wf.psi(numpy.array([[1.0,0.5,0.3],[-0.2,0.1,-0.1]]))
R=numpy.zeros((2,3),float)
R[0]=[1.0,0.3,0.2]
R[1]=[2.0,-0.2,0.1]
print "The local energy should be -1.819 and is ",wf.LocalEnergy(R)
print "The acceptance ratio of this VMC run should be about 0.323 and plot the density \
as above"
eList=VMC(wf,10000)
myStats=stats.Stats(numpy.array(eList))
print "Your statistics should be approximately (-0.885, 1.325, 0.028, 5.613)",myStats
A better acceptance ratio
Note: If you are running low on time, feel free to skip this section
30% isn't a bad acceptance ratio. It turns out we can do much better though! Recall that the acceptance ratio for Monte Carlo goes as $A(R \rightarrow R') = \min \left[\frac{\pi(R')}{\pi(R)}\frac{T(R' \rightarrow R)}{T(R \rightarrow R')} ,1 \right]$ where, in our calculation, we have that $\pi(R) = |\Psi(R)|^2$.
Now let us define that quantum force $F(R) \equiv \frac{\nabla \Psi(R)}{\Psi(R)}$. Then choose a new point $R'$ with probability $P(R') = \exp[-(R-R'-\tau F(R))^2/(4\lambda\tau)$. Notice that this will produce a non-trivial value for both $T(R \rightarrow R')$ and $T(R' \rightarrow R)$. If you implement this new Monte Carlo move, you will find that the acceptance ratio (for small $\tau$) will be approximately 1. Try modifying your VMC method to use this quantity:
Hints about implementing this:
- The biggest difficulty is implementing $\nabla \psi / \psi$. Remember that this quantity is a vector.
My code is slow :(
- Switch the numerical derivatives to analytic derivatives. Numerical derivatives are really slow!
- Do wave function updates for single particle moves
- Pythonize it: We have been writing in python but not in a very pythonic way. 'for loops' are really slow in python. On the other hand, list comprehensions and matrix operations are much faster. Find the slow parts of your code and speed them up!
- Use cython! Cython is essentially a version of python that allows you to add types and speed up python. (Some python features don't work, though).
- Well, python is slow. Maybe you should try C++ (or fortran or fortress or ocaml).
Now, we have learned how to compute integrals using Variational Monte Carlo. Often, though, we don't know what our wave function actually is (in this case, we don't know what the correct value of $\alpha$ is). In the next section, we will use the variational principle to select the best value of the paramaters. Once we've started learning how to optimize, then we can afford to make our wave function more complicated!
Continue onto the next section...